Экзокринной функции поджелудочной железы при муковисцидозе
Терминология
Муковисцидоз (кистозный фиброз, синдром Ландштайнер-Фанкони-Андерсена) — наиболее распространённое врождённое заболевание ПЖ, обусловленное мутацией гена трансмембранного регулятора кистозного фиброза (CFTR). Ген CFTR изолирован в 1989 г., содержит 27 экзонов и охватывает около 250 тыс. пар нуклеотидов, расположенных в середине длинного плеча 7-й хромосомы (7q35). В настоящее время идентифицировано более 1000 мутаций данного гена, ответственных за развития симптомов муковисцидоза.
Эпидемиология
Частота бессимптомного носительства аномального гена CFTR в популяции составляет около 3%, все мутации определяют аутосомнорецессивный тип наследования болезни. Согласно законам менделевского распределения, у родителей, гетерозиготных по гену CFTR, риск рождения ребёнка, гомозиготного по аномальному гену CFTR и больного муковисцидозом, составляет 25% и не зависит от последующих генераций.
Патоморфология
Патогенез муковисцидоза расшифрован в последние годы, когда удалось определить, что трансмембранный регулятор кистозного фиброза представляет собой регуляторный белок, расположенный в клеточных мембранах. Он состоит из 1480 аминокислот и обеспечивает регуляцию ионных каналов клетки. CFTR состоит из двух мембранных субъединиц, каждая из которых включает 6 фиксированных на мембране сегментов, двух нуклеотидсвязываюших субъединицы и цитоплазменной регуляторной субъединицы с центрами дтя фосфорилирования цАМФ-зависимой протеинкиназы А.
В результате фосфорилирования и присоединения к нуклеотидсвязывающим субъединицам CFTR двух молекул аденозинтрифосфорной кислоты белок меняет свою конформацию, открывая ионные канаты клетки. CFTR локализуется преимущественно в апикальных мембранах эпителиоцитов интралобулярных протоков экзокринных желёз и в результате своей активации обеспечивает пАМФ-зависимый транспорт ионов СТ из клетки в просвет протоков.
Вслед за транспортом ионов СГ в просвет канальцев осуществляется пассивная диффузия катионов Na2+ и воды, активный транспорт ионов Na2+ посредством Na21-, К1-АТФазы, а также обмен анионов СГ на катионы гидрокарбоната, что приводит к гидратации и защелачиванию первичного секрета экзокринных желёз.
Помимо своей основной функции регулятора С-каналов клетки CFTR играет важную роль в процессах активации Nа-каналов, трансмембранного транспорта аденозинтрифосфорной кислоты, построения цитоплазматической мембраны, эндо- и экзоцитоза, адгезии бактериальных ктеток на клеточных мембранах, регуляции функции гликозилтрансфераз внутриклеточных везикул, а также апоптоза.
Расшифровка молекулярной структуры CFTR дала возможность точно генотипировать изменения кодирующих его кодонов. Различают четыре класса мутаций CFTR.
I класс — нарушения трансляции (нонсенс, сплайсинговые мутации, сдвиг рамки считывания).
II класс — нарушения внутриклеточного транспорта CFTR (миссенс, деления).
III класс — нарушения регуляции CFTR (миссенс).
IV класс — нарушения транспортных функций CFTR (миссенс).
Все эти изменения реализуются на клеточном уровне недостаточной гидратацией и защелачиванием первичного секрета экзокринных желёз, который меняет свои физико-химические свойства. Он становится вязким и густым, в секрете увеличивается концентрация гликопротеинов, затрудняется движение его по протокам, которые обтурируются белковыми пробками и дилатирутотся. В ПЖ это приводит к тому, что, как правило, ешё до рождения ребёнка её протоки оказываются заблокированными, панкреатические ферменты не достигают ДПК, происходит их аутоактивация в ткани ПЖ с аутолизом последней.
Таким образом, для муковисиидоза патогномоничен панкреатит с выраженной экзокринной недостаточностью, которая развивается у 75% больных. Следует отметить, что у 2-15% больных функция ПЖ снижена несущественно и заболевание проявляется рецидивирующими атаками ОП.
Клиническая картина
В неонатальном периоде заболевание проявляется признаками интестинальной обструкции (мекониальный илеус) и, в ряде случаев, перитонитом, связанным с перфорацией кишечной стенки. На рентгенограмме определяют вздутие петель кишки при отсутствии уровня жидкости, при ирригоскопии выявляют микроколон. Частота ассоциации мекониального илеуса с муковисцидозом достигает 70—80%, что требует проведения потовой пробы и других диагностических мероприятий для исключения муковисцидоза всем новорождённым с мекониальным илеусом, о чём будет сказано далее. Другим проявлением муковисцидоза в неонатальном периоде выступает затяжная желтуха.
При отсутствии вышеуказанных маркёров заболевания у грудного ребёнка, больного муковисцидозом, может отмечаться стойкий сухой кашель с одышкой; обильный, зловонный, жирный стул, содержащий непереваренные остатки пищи и с трудом смывающийся с горшка, пелёнок; задержка физического развития со снижением количества подкожной жировой клетчатки и мышечной массы при нормальном или даже повышенном аппетите, гипопротеинемией и отёками. В некоторых случаях могут доминировать симптомы со стороны респираторной системы, ЖКТ или проявления нарушения нутритивного статуса и белково-энергетической недостаточности.
Однако такую типичную клиническую картину с выраженными признаками мальабсорбции и стеатореи отмечают не у всех пациентов: в некоторых случаях единственным симптомом заболевания может быть отставание в физическом развитии. Недостаточность функции ПЖ развивается в любом возрасте, но в большинстве случаев (90%) она формируется уже на первом году жизни и неуклонно прогрессирует. Для пациентов, не получающих соответствующего лечения, характерен вторичный дефицит жирорастворимых витаминов A, D, Е и К
При физикальном обследовании у детей, больных муковисцидозом, обнаруживают учащённое дыхание, увеличение переднезаднего размера грудной клетки, слабо выраженное, но стойкое втяжение нижних межрёберных мышц. В анамнезе ряда больных имеются данные о различных симптомах со стороны бронхолёгочной системы, напоминающих проявления рецидивирующих инфекций дыхательных путей, но длящихся дольше, чем у детей, не страдающих муковисцидозом, и постепенно приобретающих хроническое течение.
Аускультативные патологические признаки могут вообще не выявляться или присутствовать в виде сухих и влажных мелко- и крупнопузырчатых хрипов. На рентгенограмме органов грудной полости — уплотнение стенок бронхов, различной степени уплотнение лёгочной ткани. Могут развиваться ателектазы в сегментах и долях лёгких, причём поражение правой верхней доли относится к диагностическим признакам муковисцидоза.
Иногда родители замечают чрезмерно солёный вкус нота или кристаллики соли на коже ребёнка. Повышенное выделение солей с потом — важное клиническое проявление заболевания. Большая потеря солей через кожные покровы у детей с муковисцидозом может привести к истощению запасов натрия и хлора, хронической гипоэлсктролитемии.
Одним из нередких симптомов, настораживающих в отношении муковисцидоза в этом возрасте, выступает тепловой удар или дегидратация в жаркое время года.
Хотя у большинства больных симптомы муковисцидоза появляются уже на первом году жизни, в ряде случаев первые признаки заболевания развиваются позднее, в дошкольном возрасте. Для дошкольного возраста характерны тяжёлая недостаточность питания, нарастающие изменения стула и выпадение прямой кишки (ректальный пролапс), реже выявляют симптом «барабанных палочек», гепатомегалию с функциональными нарушениями органа.
Лишь в очень редких случаях диагноз муковисцидоза не устанавливают до достижения больными школьного возраста, что может быть связано с «мягкими» мутациями и относительной «сохранностью» функции ПЖ. При этом обычно выявляют признаки недостаточности питания, нарушения стула, персистирующис респираторные симптомы, изменения на рентгенограмме и влажные хрипы в лёгких.
К числу симптомов муковисцидоза в этом возрасте относят рецидивирующие кишечные колики, пальпируемые каловые массы и напряжение в правом нижнем квадранте живота, рвота, запоры, а также уровни жидкости при обзорной рентгенографии брюшной полости. Основной причиной указанной симптоматики выступают фекальные массы, смешанные с густым клейким секретом слизистой оболочки, которые накапливаются в форме комков в просвете кишечника, преимущественно в области слепой кишки и дистальных отделах тонкого кишечника (синдром дистальной интестинальной обструкции).
Реже абдоминальные боли выступают в качестве проявлений рецидивирующего панкреатита у детей с сохранной функцией ПЖ. К симптомам заболевания, типичным для этого возрастного периода, можно отнести хронический синусит, назальный полипоз, сахарный диабет в сочетании с респираторными симптомами. К подростковому периоду может быть обращено внимание на отставание в половом развитии.
Диагностика
Необходимость ранней диагностики муковисцидоза определена несколькими параметрами.
• Раннее начало лечения муковисцидоза обеспечивает более высокий терапевтический эффект и улучшает прогноз заболевания.
• Своевременная постановка диагноза вносит ясность в понимание родителями состояния ребёнка и позволяет им и больному вовремя адаптироваться к тяготам, связанным с хроническим заболеванием.
• Своевременная постановка диагноза позволяет семье вовремя решить необходимые вопросы, связанные с рождением здорового ребёнка (генетическое консультирование, пренатальная диагностика муковисцидоза в последующие беременности).
• Отсроченность диагноза, а следовательно, отсутствие адекватной терапии, может привести к развитию необратимых патологических изменений в легких.
• Без лечения нарушения со стороны ЖКТ могут привести к значительному отставанию в физическом развитии и недостаточности питания.
• Несвоевременность постановки диагноза может быть сопряжена с излишними, сложными, дорогостоящими диагностическими и лечебными мероприятиями, связанными с лечением осложнений заболевания.
До настоящего времени сохраняются объективные сложности при ранней диагностике заболевания, связанные как с клинической гетерогенностью проявлений основного дефекта в гене CFTP, низкой частотой распространённости большинства мутаций, нахождением их преимущественно в компаундном состоянии, так и лабильностью потового теста. Генетический полиморфизм заболевания обусловливает выраженное фенотипическое разнообразие форм заболевания от тяжёлых до субклинических. При последних хлориды пота оказываются низкими. Положительным (подтверждающим диагноз муковисцидоза) потовый тест считают, если хлор в поте превышает 60 ммоль/л. Существует пограничная зона от 40 до 60 ммоль/л. Отрицательным считают лотовый тест с хлоридами менее 40 ммоль/л.
Учитывая ситуации, когда хлориды пота имеют нормальные или пограничные значения у больных муковисцидозом, а также нередко повышение их у больных с некоторыми другими заболеваниями: синдром приобретённою иммунодефицита (СПИД), синдром Дауна, атопический дерматит, гипотиреоз, гипопаратиреоз, кахексия, психогенная анорексия, несахарный диабет, ХП, целиакия, возникает необходимость поиска более чувствительных диагностических тестов.
Один из последних тестов — измерение величины разности назальных потенциалов. Он нашёл широкое применение в экономически развитых странах. С помощью этого теста оценивают эффективность стремительно развивающейся в настоящее время генной терапии, а также новых терапевтических направлений, нацеленных на нормализацию ионных нарушений у больных муковисцидозом (стимуляция альтернативных путей секреции ионов Cl и снижение гиперабсорбции ионов Na+) К сожалению, в России этот тест проводят только в Москве и Санкт-Петербурге.
Анализ дезоксирибонуклеиновой кислоты (ДНК) при подозрении на муковисцидоз на все возможные мутации невозможен, поскольку к настоящему моменту число известных мутаций превышает 1200.
У новорождённых при подозрении на муковисцидоз необходимо определить сывороточный уровень иммунореактивного трипсина, диагностически значимый для муковисцидоза — повышение в 5-10 раз.
Важную роль в диагностике занимают тесты, направленные на оценку экзокринной функции ПЖ (копрологическое исследование, общее содержание жира в кале, определение трипсина и эластазы-1 в кале).
Лечение
В нашей стране лечение муковисцидоза проводят в специализированных учреждениях— региональных центрах муковисцидоза или в Российском центре диагностики и лечения муковисцидоза. Поскольку лечение прекрасно изложено в соответствующих руководствах, мы решили ограничить приведением только основных принципов лечения:
• лечебная физкультура;
• муколитики и бронхолитики;
• антибактериальная терапия;
• заместительная ферментная терапия панкреатическими ферментами;
• гепатопротекторы;
• витаминотерапия;
• диетотерапия;
• лечение осложнений.
Маев И.В., Кучерявый Ю.А.
Источник
Муковисцидоз (панкреофиброз, стеаторея панкреатическая врожденная и др.) — наследственная болезнь, характеризующаяся кистозным изменением поджелудочной железы, желез кишечника, дыхательных путей, больших слюнных желез и др. вследствие выделения соответствующими железами очень вязкого секрета. Наследуется по аутосомно-рецессивному типу. Считается, что 2,6-3,6% взрослого населения являются гетерозиготными носителями гена муковисцидоза.
Муковисцидоз встречается в разных регионах мира с довольно различной частотой — от 1:2800 до 1:90000 новорожденных (последняя цифра относится преимущественно к лицам монголоидных рас).
Поджелудочная железа при муковисцидозе уплотнена, избыточно развиты прослойки соединительной ткани. Выводные протоки кистозно расширены. У детей старшего возраста ацинусы расширены, отмечается кистозное расширение отдельных протоков и ацинусов — до полного кистозного превращения всей железистой паренхимы. Количество панкреатических островков такое же, как и у здоровых лиц. Развитие болезни связывают с нарушением трансмембранного транспорта ионов, что считается вызвано дефектом «кальцийзависимого регуляторного белка».
Основными симптомами муковисцидоза у взрослых являются снижение массы тела, «панкреатогенные» поносы, значительная стеаторея, постоянные легочные заболевания с формированием гнойных бронхоэктазов, компенсаторной эмфиземы легких, хронической пневмонии с часто возникающими очагами абсцедирования, наличие хронического ринита, синусита с полипозом.
Подобное сочетание довольно различных симптомов, притом наблюдаемых с детства, позволяет врачу заподозрить муковисцидоз. Рентгенологическое исследование грудной клетки и околоносовых пазух позволяет выявить изменения в них, довольно характерные для муковисцидоза. При УЗИ поджелудочной железы она может быть уплотнена, увеличена в размерах, кистозно перерождена с наличием в кистах эхонегативного содержимого. Может быть увеличенной печень. Важным методом выявления добавочной поджелудочной железы является гастродуоденоскопия, в необходимых случаях — с биопсией. Очень надежным считается так называемый потовой тест с определением в поте содержания натрия и хлора. Доказательным в пользу муковисцидоза является повышение содержания этих ионов в поте свыше 40 ммоль/л у детей и 60 ммоль/л у взрослых.
В качестве лечения муковицидоза рекомендуется бедная жирами и богатая белками диета. Рекомендуется частое (4-6 раз в день) дробное питание. Назначаются ферментные препараты, компенсирующие недостаточность внешнесекреторной функции поджелудочной железы (панкреатин, панзинорм, панцитрат, фестал, солизим, сомилаза и др.). Для разжижения густого слизистого секрета назначают ацетилцистеин (муколитический препарат). При большой потере массы тела вместе с усиленным питанием назначают анаболические стероидные гормоны. Больные должны находиться под диспансерным наблюдением гастроэнтеролога с периодическим (1 раз в 1-2 мес) проведением копрологических исследований (определяется степень нарушения переваривания, в основном это касается жиров, и соответственно подбирается доза ферментных препаратов). Больным муковисцидозом обычно рекомендуют поливитамины, особенно витамины группы В.
Больные муковисцидозом также должны находиться под диспансерным наблюдением у пульмонолога, чтобы не запустить бронхолегочный процесс, и у отоларинголога. Они должны всячески избегать переохлаждения.
 [1], [2], [3], [4], [5], [6], [7], [8], [9], [10], [11], [12], [13], [14], [15], [16], [17], [18]
[1], [2], [3], [4], [5], [6], [7], [8], [9], [10], [11], [12], [13], [14], [15], [16], [17], [18]
Источник
Для цитирования: Бельмер С.В., Коваленко А.А., Гасилина Т.В. Врожденные причины экзокринной недостаточности поджелудочной железы // РМЖ. 2004. №16. С. 984
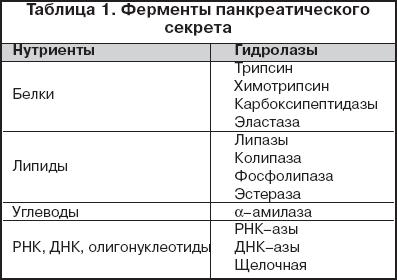
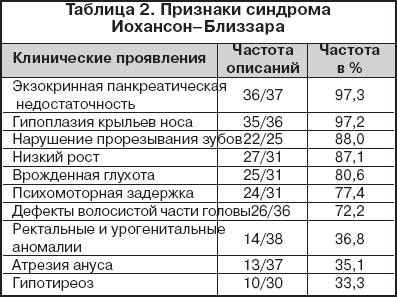
Поджелудочная железа является важнейшим экзокринным органом, обеспечивающим адекватное течение пищеварительных процессов. Экзокринная функция поджелудочной железы состоит в выработке ферментов и бикарбонатов. Экзосекреторный аппарат железы включает ацинарные клетки, образующие ацинусы, и протоки. Ацинарные клетки синтезируют и выделяют в полость ацинуса белковый секрет, 98% которого составляют ферменты. Ацинусы секретируют и электролиты (Na + , Cl – ), но в небольшом количестве. Вода и электролиты, преимущественно гидрокарбонаты, секретируются дуктулоцитами, выстилающими главные, междольковые и внутридольковые протоки, и центральными ациноцитами, образующими стенку вставочного отдела протока. Дуктулярный секрет содержит главным образом гидрокарбонат натрия, за счет которого секрет имеет основную реакцию (рН = 7,5–8,8). Функции неферментной части панкреатического секрета состоят в ощелачивании кислого желудочного содержимого, поступающего в двенадцатиперстную кишку, и, как следствие, инактивации пепсина, подавлении желудочного и стимуляции кишечного пищеварения, в обеспечении оптимального рН для гидролиза нутриентов в полости тонкой кишки, повышении активности панкреатических и кишечных гидролаз, которые гидролизуют практически все макронутриенты (табл. 1). Переваривание белковых субстратов осуществляется протеазами. Трипсин, химотрипсин и карбоксипептидаза вырабатываются экзокринными клетками поджелудочной железы в виде функционально неактивных предшественников – трипсиногена, химотрипсиногена и прокарбоксипептидазы. Трипсин и химотрипсин расщепляют полипептиды, образовавшиеся в желудке под действием пепсина. Деградация коротких пептидов осуществляется карбоксипептидазой и аминопептидазой, секретируемой в тонкой кишке. Превращение трипсиногена в трипсин происходит в тонкой кишке под действием протеолитического фермента энтерокиназы, секретируемой клетками кишечного эпителия путем отщепления гексапептида от N–конца полипептидной цепи. Свободный трипсин гидролизует пептидные связи, образованные с участием лизина и аргинина. Химотрипсиноген превращается в химотрипсин под действием трипсина и гидролизует пептидные связи, образованные остатками фенилаланина, тирозина и триптофана. Специфичность протеаз поджелудочной железы по отношению к пептидным связям разных аминокислот обеспечивает очень высокую эффективность переваривания белков. Карбоксипептидаза – цинксодержащий фермент, синтезируемый в виде предшественника прокарбоксипептидазы. В активном состоянии карбоксипептидаза последовательно отщепляет от пептидов С–концевые остатки. N–концевые остатки отщепляются под действием аминопептидазы. Проэластаза под действием трипсина превращается в эластазу, которая вызывает деградацию волокон эластина и некоторых других белков. В результате последовательного действия протеолитических ферментов и пептидаз перевариваемые белки превращаются в смесь свободных аминокислот, транспортируемых через эпителий тонкой кишки. Переваривание триглицеридов осуществляется под действием липазы в присутствии желчных кислот и колипазы. Фермент синтезируется ациноцитами в активном состоянии, не вызывая лизис клеток, поскольку функционирует исключительно в отношении эмульгированных жиров. Активность липазы повышают ионы кальция, хлористый натрий. Колипаза способствует абсорбции липазы на слизистой оболочке тонкой кишки, повышая ее активность в зоне щеточной каемки. Активная липаза гидролизует один или оба крайних жирнокислотных остатка с образованием смеси свободных жирных кислот в виде их натрий–калиевых солей (мыл), ди– и моноглицеридов, глицерина. Фосфолипазы гидролизуют фосфолипиды. Они вырабатываются в неактивной форме и активируются трипсином. Гидролиз углеводов протекает под действием ? –амилазы, активность которой зависит от присутствия в среде ионов хлора. Экзокринная недостаточность поджелудочной железы может развиваться в результате врожденных и приобретенных причин. Значительная недостаточность экзокринной функции поджелудочной железы с выпадением преимущественно липазной активности проявляется непереваренным частым, обильным стулом с характерным жирным блеском и своеобразным запахом. В то же время умеренная или незначительная панкреатическая недостаточность часто выявляется лишь при проведении специального обследования. Простейшим тестом на экзокринную панкреатическую недостаточность является увеличение нейтрального жира в копрограмме. В современной практике широко используется липидограмма кала, позволяющая получить количественную оценку стеатореи, а также определение в кале эластазы–1, протеолитического фермента поджелудочной железы, на уровень которого не влияют ни диета больного, ни прием препаратов заместительной терапии. Причинами экзокринной панкреатической недостаточности могут быть врожденные аномалии поджелудочной железы, наблюдающиеся относительно редко и часто протекающие бессимптомно. Впервые поджелудочная железа выявляется у 4–х–недельного эмбриона в виде двух выпячиваний первичной кишки дистальнее желудка. Дорзальная почка быстро удлиняется и в конечном итоге формирует хвост, тело, и часть головки будущей поджелудочной железы. Вентральная почка соединяется с желчевыводящим протоками и в дальнейшем формирует головку поджелудочной железы. В течение последующих недель происходит позиционирование двенадцатиперстной кишки, билиарной системы и поджелудочной железы. Приблизительно в конце шестой недели гестации две части поджелудочной железы соединяются. Вентральный проток открывается в фатеров сосок вместе с общим желчным протоком и представляет собой основной проток поджелудочной железы (проток Wirsung). Дорсальный проток формирует вспомогательный проток (проток Santorini ), сохраняющийся функционально активным у 70% взрослых лиц. Основными аномалиями поджелудочной железы являются гипоплазия, дисплазия, аномалии протоков, разделенная поджелудочная железа ( pancreas divisum ), кисты холедоха, гетеротопия поджелудочной железы, кольцевидная поджелудочная железа. Полная агенезия поджелудочной железы встречается редко и несовместима с жизнью. При парциальной агенезии поджелудочная железа уменьшена в размере и/или имеет дефектную форму, обычно связана с аномальным развитием вентрального зачатка органа. При гипоплазии поджелудочная железа имеет нормальные размеры и форму, однако эпителиальные клетки замещены жировой тканью, система протоков редуцирована, а их терминальные отделы слабо дифференцированы. Дисплазия представляет собой дезорганизацию паренхимы, протоков и избыток фиброзной и мышечной ткани. Клинически перечисленные выше аномалии могут проявляться стеатореей, а также гипергликемическими состояниями. В то же время нередки и случаи бессимптомного течения, если частично сохранившаяся функция органа оказывается достаточной. В отдельных случаях наблюдаются задержка внутриутробного развития, а также другие аномалии развития. Выявить аномалию поджелудочной железы помогают ультразвуковое исследование, компьютерная и магнитно–резонансная томография, а также эндоскопическая ретроградная пакреатохоланиография. Различные варианты формирования протоков поджелудочной железы наблюдаются примерно у 30–40% лиц и в большинстве случаев ничем себя не проявляют. В то же время аномалии панкреатических протоков могут стать причиной развития панкреатита, а также проявляться экзокринной панкреатической недостаточностью. Pancreas divisum является одной из наиболее часто выявляемых аномалий поджелудочной железы: она регистрируется примерно в 7,5% случаев от всех проведенных ретроградных панкреатохолангиографий, а также в 50% случаев ретроградных панкреатохолангиографий, проведенных в связи с диагностированным панкреатитом. Аномалия развивается вследствие нарушенного слияния двух зачатков поджелудочной железы. В результате отток от большей части органа осуществляется по относительно узкому санториниеву протоку, что приводит к повышению давления в ацинусах и развитию панкреатита. Естественно, что нарушение оттока панкреатического секрета приводит к более или менее выраженной мальабсорбции. В соответствии с классификацией Alonso–Lej и соавт. выделяют три основных типа кист холедоха: (1) дилатация всего протока, (2) саккуларные дилатации части протока, и (3) холедохоцеле, кистозное расширение интрадуоденальной части протока. Кисты холедоха, аномальная длина и аномальное слияния панкреатического и общего желчного протоков чаще ассоциируются с развитием панкреатита и холестазом и в меньшей степени – со стеатореей, хотя мальабсорбция в том или ином виде присутствует практически во всех случаях. Основным методом диагностики выявления перечисленных выше аномалий является эндоскопическая ретроградная панкреатохолангиография. Хотя гетеротопированная поджелудочная железа может проявляться самой разнообразной симптоматикой (болями в животе, кишечными кровотечениями, рвотами и др.), в большинстве случаев она оказывается случайной находкой и крайне редко сопровождается панкреатической недостаточностью. Последнее справедливо и в отношении кольцевидной поджелудочной железы, клинические проявления которой обусловлены сдавлением двенадцатиперстной кишки и нарушением кишечного пассажа и/или развитием панкреатита. Наиболее тяжелые случаи экзокринной панкреатической недостаточности врожденного происхождения связаны с муковисцидозом, синдромом Швахмана–Даймонда ( Schwachman – Diamond syndrome ), синдромом Иохансон–Близзара ( Johanson – Blizzard syndrome), аплазией/гипоплазией поджелудочной железы. Также описаны варианты изолированных дефицитов панкреатических ферментов: липазы, липазы–колипазы, колипазы, амилазы, трипсиногена. Муковисцидоз достаточно хорошо описан в современной литературе и, к сожалению, не является редкой патологией. Напротив, это одно из наиболее распространенных генетических заболеваний. В нашей стране его распространенность составляет 1:12 000 новорожденных. Муковисцидоз представляет собой частое моногенное заболевание, обусловленное мутацией гена МВТР (трансмембранного регулятора муковисцидоза), характеризующееся поражением экзокринных желез жизненноважных органов и систем и имеющее обычно тяжелое течение и прогноз. Заболевание наследуется по аутосомно–рецессивномутипу. При муковисцидозе имеется характерный для всех эпителиальных клеток организма дефект секреции, первично – для хлоридных ионов со вторичным снижением общего объема секреции. Определяющими для жизни больного являются характер и степень поражения легких, а также желудочно–кишечного тракта, прежде всего – поджелудочной железы и печени. Диагноз в настоящее время базируется на наличии хронического бpонхолегочного процесса, типичного кишечного синдрома (стеаторея), случаев муковисцидоза у сибсов и положительного потового теста. Патогномоничным остается потовый тест, который поводится не менее трех раз методом пилокаpпинового электрофореза. Пpи муковисцидозе содержание натрия и хлора в потовой жидкости превышает 60 ммоль/л, при этом навеска пота должна быть не менее 100 мг. Пpи получении пограничных значений хлоридов пота (40–60 ммоль/л) необходимо проводить ДНК–анализ. Синдром Швахмана–Даймонда, описанный впервые в 1964 году, встречается с частотой 1:10 000–1:20 000 живых новорожденных. Это врожденное заболевание характеризуется панкреатической недостаточностью (преимущественно – липазной) на фоне гипоплазии поджелудочной железы, гематологическими сдвигами (чаще – нейтропенией, но также могут наблюдаться анемия и тромбоцитопения), задержкой роста, костными аномалиями (метафизарная дисхондроплазия, чаще поражаются головки бедренных костей и коленные суставы, возможны клинодактилия, гипоплазия фаланг, узкая грудная клетка). Клиническая картина полиморфна и зависит от преобладающего синдрома. Нарушение функции костного мозга приводит к развитию иммунодефицитного состояния и рецидивирующим инфекциям. Т.к. в настоящее время заместительная терапия даже тяжелой панкреатической недостаточности достаточно хорошо отработана, прогноз заболевания определяется в большей степени выраженностью гематологических сдвигов, особенно нейтропении, и как следствие – частотой инфекционных осложнений. Синдром Иохансон–Близзара был впервые описан в 1971 г. Он включает в себя экзокринную панкреатическую недостаточность, гипоплазию крыльев носа, нарушение прорезывания зубов, задержку роста, врожденную глухоту, задержку психомоторного развития, эктодермальные дефекты волосистой части головы и многие другие (табл. 2). Заболевание передается по аутосомно–рецессивному типу. Врожденная липазная недостаточность (синдром Sheldon–Rey, описанный в 1964 году) проявляет себя с рождения учащенным жирным стулом и всеми соответствующими лабораторными признаками. Сложность диагностики этого заболевания связана с необходимостью исключить все прочие заболевания, проявляющиеся панкреатической недостаточностью. В случае адекватной коррекции нарушенной функции поджелудочной железы высокоактивными препаратами панкреатических ферментов прогноз относительно благоприятный. Описано также три случая сочетания недостаточности липазы и колипазы, а также два случая (у двух братьев) изолированной недостаточности липазы. Методы коррекции Коррекция экзокринной панкреатической недостаточности определяется степенью ее выраженности. При тяжелой врожденной недостаточности (муковисцидоз, синдром Швахмана–Даймонда, врожденная липазная недостаточность) назначается диета с некоторым повышением калорийности, содержания белка и физиологическим содержанием жира. Также необходимо адекватное состоянию больного включение в питание (и терапию) витаминов (особенно жирорастворимых), микро– и макроэлементов. Заместительная терапия, направленная на коррекцию сниженной экзокринной функции поджелудочной железы, должна проводиться современными микросферическими препаратами панкреатических ферментов с рН–чувствительной оболочкой. Высокая активность этих препаратов обусловлена высокой степенью активности исходного субстрата (панкреатина), особой формой препарата, которая обеспечивает его равномерное перемешивание с желудочным содержимым и синхронное прохождение в двенадцатиперстную кишку и рН–чувствительной оболочкой микросфер, защищающей ферменты от разрушения в желудке и обеспечивающей их высвобождение в двенадцатиперстной кишке. Сами микросферы помещены в рН–чувствительные капсулы для защиты от преждевременной активации в ротовой полости и в пищеводе (где также, как и в двенадцатиперстной кишке имеет место щелочная среда) и облегчения приема препарата. Таким образом, препарат достигает желудка, где капсулы растворяются, а микросферы высвобождаются и перемешиваются с желудочным содержимым. В двенадцатиперстной кишке при значения рН около 5,5 рН–чувствительная оболочка микросфер растворяется и высокоактивные ферменты начинают свое действие. Микросферический препарат Креон 10 000 (Solvay Pharma, Германия) в 1 капсуле содержит 10 000 Ед липазы, 8000 Ед амилазы и 600 Ед протеаз, а более активная форма Креона – 25 000 (25 000 Ед, 18 000 Ед и 1000 Ед соответственно). Клинические наблюдения позволяют сделать заключение о его значительной клинической эффективности и полной безопасности, даже при непрерывном, длительном (многолетнем) применении у детей. Более того, назначение высокоактивных препаратов панкреатических ферментов позволяет сохранить возрастное содержание липидов в диете (отказаться от их ограничения) и существенно повысить качество жизни. Доза препарата подбирается индивидуально, с учетом степени панкреатической недостаточности. В тяжелых случаях (например, при муковисцидозе) суточное количество капсул препарата может быть достаточно большим (10–20 и более). Контроль за адекватностью заместительной терапии проводится не только клинически, но также лабораторно (копрограмма, липидограмма кала). Таким образом, экзокринная недостаточность поджелудочной железы может быть связана с разнообразными врожденными заболеваниями. Ранняя их диагностика позволяет своевременно назначить заместительную терапию, которая во многих случаях определяет прогноз не только заболевания, но и жизни пациента.
Поделитесь статьей в социальных сетях
Источник
